 Loading... Please wait...
Loading... Please wait...- Call us on 443-686-9618
- Wish Lists
- My Account
- 0.00
Categories
- Home
- IHC Detection System
- DAB (SA-HRP) TUNEL Cell Apoptosis Detection Kit
 TUNEL Cell Apoptosis Detection Kit")
DAB (SA-HRP) TUNEL Cell Apoptosis Detection Kit

 TUNEL Cell Apoptosis Detection Kit, $399.00 (https://store.ihcworld.com/dab-sa-hrp-tunel-cell-apoptosis-detection-kit/))
Product Description
Descirption/Introduction
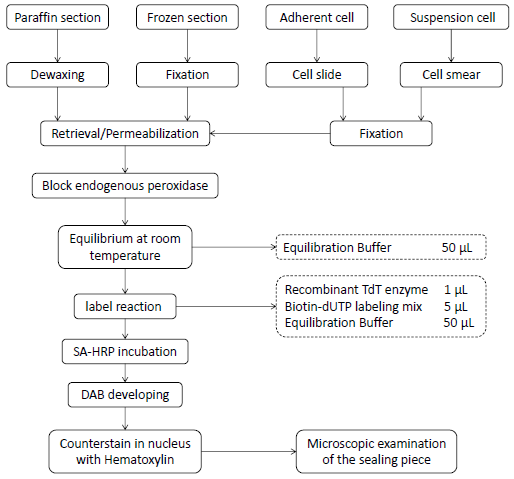
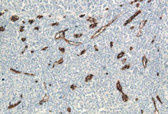
The breakage of chromosomal DNA in apoptosis is a gradual process. Chromosomal DNA is first degraded into large fragments of 50-300 KB under the action of endogenous nuclease hydrolase, and then about 30% of chromosomal DNA is randomly cut between nucleosome units under the action of ca2+ and mg2+ dependent endonuclease to form 180-200 BP nucleosome DNA polymer. Therefore, in the late stage of apoptosis, DNA will be degraded into 180-200 BP fragments, and a large number of 3'-oh terminals are exposed on the broken genomic DNA. Terminal deoxyribonucleotidyl transferase (TDT) is a template independent DNA polymerase, which can catalyze the binding of deoxynucleotides to the 3'-oh terminal of broken DNA molecules. Therefore, TUNEL (TDT mediated dUTP nick end labeling) cell apoptosis detection kit can be used to detect the nuclear DNA breakage of tissue cells in the late process of apoptosis. The principle is that under the action of TDT enzyme, biotin labeled dUTP (biotin dUTP) is added to the 3´-OH terminal exposed during genomic DNA breakage, and then streptavidin (SA HRP) labeled with horseradish peroxidase (HRP) is used to detect the biotin labeled DNA terminal. Finally, the color reaction is carried out by adding the substrate mixture (DAB) of HRP, the nuclei of apoptotic cells were stained brown, which could be detected by general optical microscope. The kit has a wide range of applications, and is suitable for the detection of apoptosis in paraffin tissue sections, frozen tissue sections, cell climbing slides, cell smears, etc.
Storage and Handling Conditions
Wet ice transportation; Stored at -20 ℃ , valid for 12 months.
Component
|
Component Number |
Component |
G1507-50T |
G1507-100T |
|
1507-1 |
Recombinant TdT Enzyme |
50 μL |
2×50 μL |
|
1507-2 |
Biotin-dUTP Labeling Mix |
250 μL |
2×250 μL |
|
1507-3 |
Equilibration Buffer |
5x1 mL |
10×1 mL |
|
1507-4 |
Streptavidin-HRP |
25 μL |
2×25 μL |
|
1507-5 |
Proteinase K(200 µg/mL) |
1 mL |
2×1 mL |
|
Manual |
1 pc |
||
Preparation
1. PBS phosphate buffer (recommended G0002).
2. Stationary solution: 4% paraformaldehyde dissolved in PBS, pH 7.4 (recommended G1101).
3. Membrane breaker: 0.1%-0.5% Triton X-100 (G1204 recommended).
4. Hydrogen peroxide sealing solution: 3% H2O2, prepared with PBS.
5. If you need to dye nuclei, you need to prepare hematoxylin dye solution (recommended G1004) and hematoxylin differentiation solution (recommended G0139) Hematoxylin bluing solution (recommended G1004)
6. Self prepared DAB color reagent (recommended G1212), n-butanol, xylene, neutral gum, etc
7. Please wear test clothes and disposable gloves during operation.
Assay Protocol / Procedures
1. Sample preparation
A. Paraffin embedded tissue section
1) Paraffin tissue sections were immersed in xylene for 5-10 min at room temperature and repeated 3 times; Then soak in absolute ethanol for 5 min and repeat twice; Finally, it was immersed in gradient ethanol (85%, 75%) and double-distilled water once each for 5 min;
2) Gently moisten the slices with PBS and remove the excess liquid around the sample. Use a PAP Pen to draw a small circle 2-3 mm apart from the tissue along the peripheral contour of the tissue, so as to facilitate downstream permeability treatment and balance marking. During the experiment, do not let the sample dry, and put the treated sample in the wet box to keep the sample moist;
3) Prepare Proteinase K working solution: dilute Proteinase K (200 µg/ml) stock solution with PBS as diluent at the volume ratio of 1:9, so that the final concentration is 20 μg/mL;
4) Add 100 μL Proteinase K working solution drops to each sample, completely covered the tissue and incubated at 37℃ for 20 min;
Note: Proteinase K treatment is mainly helpful for the permeability of staining reagents in subsequent steps of tissues and cells. Too long or too short incubation time will affect the subsequent labeling efficiency. In order to obtain better results, the incubation time of Proteinase K can be optimized.
5) Soak and clean the sample with PBS solution for 3 times, each time for 5 min (Proteinase K needs to be cleaned, otherwise it will interfere with the subsequent marking reaction). The treated sample is placed in a wet box to keep the sample moist;
6) (optional steps) remove the excess liquid from the sample, add an appropriate amount of membrane breaking liquid to the tissue, fully infiltrate the tissue, and treat at room temperature for 20 min; After the membrane breaking treatment is completed, the sample shall be moistened with PBS solution for 3 times for 5 min each time; The treated sample is placed in a wet box to keep the sample moist;
7) Remove the excess liquid from the sample, drop an appropriate amount of 3% H2O2 (prepared with PBS) onto the tissue, fully infiltrate the tissue, and treat it at room temperature for 20 min (inactivate the endogenous peroxidase in the tissue, and the incubation time should not be too long, otherwise DNA breakage caused by hydrogen peroxide will occur, resulting in false positive); Then moisten the sample with PBS solution for 3 times, each time for 5 min; The treated sample is placed in a wet box to keep the sample moist.
B. Tissue frozen section
1) Immerse the slide in 4% paraformaldehyde solution (dissolved in PBS) for fixation, and incubate at room temperature for 10-15 min;
2) After the film is removed from the fixing liquid, it shall be naturally dried in the fume hood;
3) Put the slide into pure water or PBS for moistening and washing, and remove the residual fixed liquid on the slide;
4) Draw a small circle 2-3 mm apart from the tissue along the peripheral contour of the tissue with a PAP Pen to facilitate downstream permeability treatment and balance marking; During the experiment, do not let the sample dry, and put the treated sample in the wet box to keep the sample moist;
5) Prepare Proteinase K working solution: dilute Proteinase K (200 µg/ml) stock solution with PBS as diluent at the volume ratio of 1:9, so that the final concentration is 20 μg/mL;
6) Add 100 μL Proteinase K working solution drops to each sample, completely covered the tissue and incubated at 37 ℃ for 20 min;
Note: Proteinase K treatment is mainly helpful for the permeability of staining reagents in subsequent steps of tissues and cells. Too long or too short incubation time will affect the subsequent labeling efficiency. In order to obtain better results, the incubation time of Proteinase K can be optimized.
7) Moisten the sample with PBS solution for 2-3 times to remove the excess liquid (Proteinase K needs to be cleaned, otherwise it will interfere with the subsequent marking reaction). The treated sample is placed in a wet box to keep the sample moist;
8) (optional steps) add an appropriate amount of membrane breaking liquid to the tissue, fully infiltrate the tissue, and treat it at room temperature for 20 min. after the membrane breaking treatment, similarly wash the sample with PBS solution to remove the excess liquid. The treated sample is placed in a wet box to keep the sample moist;
9) Remove the excess liquid from the sample, drop an appropriate amount of 3% H2O2 (prepared with PBS) onto the tissue, fully infiltrate the tissue, and treat it at room temperature for 20 min (inactivate the endogenous peroxidase in the tissue, and the incubation time is not easy to be too long, otherwise DNA breakage caused by hydrogen peroxide will occur, resulting in false positive); Then moisten the sample with PBS solution for 3 times, each time for 5 min; The treated sample is placed in a wet box to keep the sample moist.
C. Cell climbing sheet
1) Adherent cells were cultured on lab Tek. After apoptosis induction treatment, the slides were gently rinsed twice with PBS;
2) Add an appropriate amount of 4% paraformaldehyde solution (dissolved in PBS) to each slide chamber for fixation, and incubate at room temperature for 20 min;
3) Remove the stationary solution and add PBS for cleaning for 3 times, each time for 5 min;
4) Each sample was immersed in membrane breaking solution and incubated at room temperature for 5 min for permeability treatment(Note: It is recommended to use 2-20 μg/mL of Proteinase K working solution for digestion, and treat at 37℃ for about 10 min, depending on the cell status. If the cells are prone to drop off, it is recommended to treat them with membrane-breaking solution);
5) Immerse the cleaning sample in an open beaker containing PBS solution for 2-3 times; Add an appropriate amount of 3% H2O2 (prepared with PBS) to each slide chamber and treat it at room temperature for 20 min;
6) Gently remove the excess liquid, add PBS for cleaning for 3 times, 5 min each time; Carefully dry the liquid around the sample on the slide with filter paper. The treated sample is placed in a wet box to keep the sample moist.
D. Cell smear
1) Cells were resuspended in PBS at a concentration of approximately 2×107 cells/mL, and 50-100 μL of cell suspension was aspirated onto an anti-debonding slide, and a clean slide was used to gently spread the cell suspension;
2) Immerse the glass slides in a staining tank containing 4% of the fresh paraformaldehyde prepared in PBS, fix the cells, and place them at 4 ℃ for 25 min;
3) Immerse the slide in PBS, place it at room temperature for 5 min, and wash it again;
4) Each sample was immersed in membrane breaking solution and incubated at room temperature for 5 min for permeability treatment (Note: It is recommended to use 2-20 μg/mL of Proteinase K working solution for digestion, and treat at 37℃ for about 10 min, depending on the cell status. If the cells are easy to fall off, it is recommended to use the membrane-breaking solution for treatment);
5) Immerse the cleaning sample in an open beaker containing PBS solution for 2-3 times; Add an appropriate amount of 3% H2O2 (prepared with PBS) to each sample and treat it at room temperature for 20 min;
6) Gently remove the excess liquid, add PBS for cleaning for 3 times, 5 min each time; Finally, carefully dry the liquid around the sample on the slide with filter paper. The treated sample is placed in a wet box to keep the sample moist.
2. DNase I treatment positive control experiment (optional steps)
After the sample permeability treatment, treat the sample with DNase I (recommended G3342) to prepare the positive control.
1) Add 100 μL 1 × DNase I buffer (preparation method: take 10 μL 10 × DNase I buffer, and then add 90 μL deionized water mixed evenly) to the permeable sample and incubated at room temperature for 5 min;
2) Gently remove excess liquid and add 100 μL working liquid containing DNase I (20 U/ml), incubate at room temperature for 10 min;
3) Gently remove the excess liquid, and thoroughly wash the slide 3-4 times in the staining tank with PBS;
(Note: the positive control slide must use a separate staining cylinder, otherwise the residual DNase I on
the positive control slide may introduce a high background on the experimental slide).
3. Marking and testing
1) Balance: add 50 μL equip buffer drops per sample to cover all the sample areas to be tested, and incubate at room temperature for 10 min;
2) Preparation of labeling solution: thaw the biotin dUTP labeling mix and equivalence buffer on ice, and mix sufficient TDT incubation buffer for all experiments according to the proportion of recombinant TDT enzyme:biotin dUTP labeling mix:equivalence buffer=1 µL:5 µLl:50 µL (1:5:50). The volume of the reagent used in the specific experiment can be adjusted according to the size of the slide;
3) Negative control system: prepare a control TDT incubation buffer without recombinant TDT enzyme and replace it with ddH2O;
4) Marking: try to remove the balanced equalization buffer, and then add 56 μL TDT incubation buffer to each tissue sample, incubated at 37 ℃ for 1 h; Be careful not to dry the slide, and keep the slide away from light;
5) Immediately moisten and wash the tissue samples with PBS for 4 times, each time for 5 min;
6) Gently wipe the PBS solution around the sample with filter paper;
7) Streptavidin HRP reaction: after drying the slides, each sample tissue was added with 100 µL (infiltrated tissue) streptavidin HRP reaction solution (streptavidin-HRP:TBST=1:200-500 dilution in advance), and incubated at 37 ℃ for 30 min;
8) Clean the sample with PBS for 3 times, 5 min each time;
9) DAB color development: prepare DAB color development working solution (prepared and used now), G1212 is recommended. Add 50-100 µL DAB color developing working solution to each sample, put the slices under the microscope to observe the color development in real time, put the slices into the wet box immediately after the positive display, and wash them with pure water to terminate the reaction;
10) Hematoxylin staining nucleus: after the sections are stained with hematoxylin staining solution for 3-5 min, they are immediately washed with pure water. After differentiation with hematoxylin differentiation solution for about 2 s, they are immediately washed with pure water, and then returned to blue with hematoxylin re blue solution for a few seconds. After the staining nucleus is completed, they need to be examined under the microscope. If the staining is too deep, they return to the differentiation solution for re differentiation; if the staining is too light, they can be re stained from the hematoxylin staining nucleus;
11) Transparent: the sample is dehydrated with fresh absolute ethanol for 4 times, each time for 5 min; Soak it in n-butanol for 5 min, put it into xylene for 5 min to be transparent, and replace it with fresh xylene for 5 min to be transparent again;
12) Sealing: use neutral gum to seal the slices and dry them naturally or in an oven at 60 ℃;
13) Microscopic examination: the samples were examined with a white light microscope (the apoptotic positive nuclei were dyed brown, and the normal negative nuclei were dyed blue).
4. Experiment flow diagram