 Loading... Please wait...
Loading... Please wait...- Call us on 443-686-9618
- Wish Lists
- My Account
- 0.00
Categories
- Home
- IHC Detection System
- Click-iT 488 TUNEL Cell Apoptosis Detection Kit

Click-iT 488 TUNEL Cell Apoptosis Detection Kit

)
Product Description
Product Description/Introduction
The breaking of chromosomal DNA in apoptosis is a gradual, phased process. Chromosomal DNA is first degraded into large 50-300 kb segments by endogenous nucleic acid hydrolases, and then about 30% of chromosomal DNA is randomly cut between nucleosome units in the presence of Ca2+- and Mg2+-dependent nucleic acid endonucleases to form 180-200 bp nucleosomal DNA multimers.Thus, in late apoptosis, DNA is degraded into 180-200 bp fragments, and a large number of 3'-OH ends are exposed on the broken genomic DNA. Terminal Deoxynucleotidyl Transferase (TdT) is a template-independent DNA polymerase that catalyzes the incorporation of deoxyribonucleotides into the 3'-OH ends of broken DNA molecules.Therefore TUNEL (TdT mediated dUTP Nick End Labeling) Cell Apoptosis Detection Kit can be used to detect the breakage of nuclear DNA in tissue cells during late apoptosis.
The principle is that under the action of TdT enzyme, EdUTP (a dUTP with alkyne modification) is doped into the 3´-OH end exposed during genomic DNA breakage, and the alkyne group reacts with the azide dye in a ring-forming reaction catalyzed by a monovalent copper ion (click reaction) thus introducing the fluorescent moiety in a targeted way. iF488 azide is used in this kit, thus it can be detected by fluorescence microscope or flow cytometry (iF488 excitation 491 nm, emission 516 nm). Compared to other modified dUTPs, EdUTP has a smaller spatial site resistance and is more easily doped into DNA ends by TdTase.
This kit has a wide range of applications and is suitable for apoptosis detection in paraffin tissue sections, frozen tissue sections, cell crawls and cell smears.
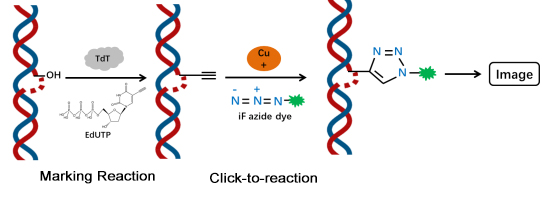
Figure 1. Schematic diagram of the click chemistry-based TUNEL kit principle
Storage and Shipping Conditions
Ship with wet ice; Store at -20℃ for 12 months.
Product Content
|
Component Number |
Component |
G1506-50T |
|
G1506-1 |
Recombinant Tdt Enzyme |
50 μL |
|
G1506-2 |
EdUTP Labeling Mix |
250 μL |
|
G1506-3 |
Equilibration Buffer |
5×1 mL |
|
G1506-4 |
Proteinase K (200μg/mL) |
1 mL |
|
G1506-5 |
iF488 azide dye |
80 μL |
|
G1506-6 |
Reaction Buffer A |
5×1 mL |
|
G1506-7 |
Reaction Buffer B |
60 μL |
|
G1506-8 |
Reaction additive (Reagent C) |
2×100 mg |
|
Manual |
1 pc |
|
Pre-experimentation
1. PBS phosphate buffer (recommended G0002 or G4202);
2. Fixed solution: 4% paraformaldehyde dissolved in PBS, pH 7.4 (recommended G1101);
3. Membrane-breaking solution: 0.1% Triton X-100 dissolved in 0.1% sodium citrate (recommended G1204);
4. 0.2% Triton X-100 prepared in PBS; 0.1% Triton X-100 prepared in PBS containing 5 mg/mL BSA;
5. For nuclear staining, provide your own DAPI (2 µg/mL), Hoechst 33258, or PI (1 µg/mL) (G1012, G1011, G1021 recommended);
6. DNase I (G3342) for positive control experiments;
7. Reaction additive (Reagent C) is centrifuged at low speed, 100 mg of the powder is dissolved in 1 mL of ultrapure water (ready to use), and 100 μL is dispensed and stored at -20°C, leaving the remaining powder for spare use; (Reagent C is easy to oxidize, try to avoid long time exposure to the air, after formulated into an aqueous solution, it is strongly recommended to divide into small portions for use; After testing the aqueous solution of Reagent C, the color of a slight change, click the reaction system can still be carried out normally, and if it shows brown color, it indicates that the component has been invalidated)
8. If the background color of the result is too dark, it may be caused by insufficient washing and residual fixative during the experiment;
9. Be careful to add the reaction liquid in sequence, and mix well while adding;
10. For your health and safety, please wear lab coat and gloves during operation.
Assay Protocol / Procedures
1. Sample Preparation
A. Paraffin-embedded tissue sections
a. The paraffin tissue sections were immersed in Eco-friendly dewaxing clear solution (G1128) for 5-10 min at room temperature and repeated 3 times. Then soak in anhydrous ethanol for 5 min and repeat twice; Finally, soak in gradient ethanol (85%, 75%, and double-distilled water) once for 5 min each;
b. Gently moisten the section with PBS and remove excess liquid around the sample, use a PAP Pen to draw a small circle spaced 2-3 mm apart from the tissue along the tissue's peripheral contour to facilitate downstream permeability processing and equilibrium labeling operations, do not allow the sample to dry out during experiments, and keep the processed samples in a wet box to keep the samples moist;
c. Preparation of Proteinase K working solution: Dilute the Proteinase K (200 µg/mL) stock solution with PBS as diluent at a ratio of 1:9 to give a final concentration of 20 µg/mL;
d. Add 100 μL of the above Proteinase K working solution to each sample to make it fully covered, and incubate at 37°C for 20 min;
Note: Proteinase K treatment mainly contributes to the permeation of staining reagents in the subsequent steps of tissues and cells, and its incubation time is too long or too short to affect the efficiency of the subsequent labeling, in order to get better results, the incubation time can be optimized according to the actual situation.
e. Wash the samples by moistening with PBS solution 3 times for 5 min each time (Proteinase K needs to be washed clean or it will interfere with the subsequent labeling reaction). The treated samples were placed in a wet box to keep the samples moist;
f. (Optional step) Remove the excess liquid on the sample, add appropriate amount of membrane-breaking solution droplets to the tissue, fully infiltrate the tissue, and treat it for 20 min at room temperature; after the membrane-breaking treatment is completed, the sample is similarly washed with PBS solution for 3 times, each time for 5 min; the treated sample is placed in a wet box to keep the sample moist.
B. Tissue frozen section
a. The tissue frozen sections are submerged in the fixative and incubated and fixed for 10-15 min at room temperature;
b. The tissue sections are removed from the fixative and placed in a fume hood to dry naturally;
c. Tissue sections are moistened and washed in purified water or PBS to remove residual fixative from the sample;
d. Use a PAP Pen to draw a small circle spaced 2-3 mm apart from the tissue along the tissue's peripheral contour to facilitate downstream permeability processing and equilibrium labeling operations, do not allow the sample to dry out during experiments, and keep the processed samples in a wet box to keep the samples moist;
e. Preparation of Proteinase K working solution: Dilute the Proteinase K (200 µg/mL) stock solution with PBS as diluent at a ratio of 1:9 to give a final concentration of 20 µg/mL;
f. Add 100 μL of the above Proteinase K working solution to each sample to make it fully covered, and incubate for 10 min at room temperature;
Note: Proteinase K treatment mainly contributes to the permeation of staining reagents in the subsequent steps of tissues and cells, and its incubation time is too long or too short to affect the efficiency of the subsequent labeling, in order to get better results, the incubation time can be optimized according to the actual situation.
g. Wash the sample 2-3 times with PBS solution to remove excess liquid (Proteinase K needs to be washed clean or it will interfere with the subsequent labeling reaction), and keep the treated sample in a wet box to keep the sample moist;
h. (Optional step) Add appropriate amount of membrane-breaking solution droplets to the tissue, fully infiltrate the tissue, room temperature treatment for 20 min, membrane-breaking treatment is completed the same after the completion of the sample with the PBS solution to rinse, to remove the excess liquid, after the treatment of the sample in the wet box to keep the sample wet.
C. Cell creep
a. Adherent cells are cultured on Lab-Tek slide chambers (Chamber Slides), and slides are gently moistened and washed 2 times with PBS after apoptosis induction treatment;
b. Appropriate amount of fixative is added to each slide chamber to cover the tissue and incubated for 20 min at room temperature;
c. Remove the fixative and add PBS to wash 3 times for 5 min each;
d. Each sample is immersed in 0.2% Triton X-100 solution prepared in PBS and incubated for 5 min at room temperature for permeabilization;
e. Wash samples by submerging them 2-3 times in an open beaker containing PBS solution;
f. Gently remove excess liquid and use filter paper to carefully blot the liquid around the sample on the slide.The processed sample is placed in a wet box to keep the sample moist.
D. Cell smear
a. Cells are resuspended in PBS at a concentration of about 2 × 107 cells/mL, and 50-100 μL of cell suspension is aspirated onto an anti-detachment slide, and a clean slide is used to gently spread the cell suspension;
b. The cell smears are immersed in a staining jar filled with fixing solution, and the cells are fixed and left at 4°C for 25 min;
c. Immerse the slide in PBS and leave it at room temperature for 5 min to soak and rinse, repeat once;
d. Gently remove the excess liquid and carefully blot the excess liquid around the sample on the slide with filter paper, draw a small circle along the peripheral contour of the cell with a PAP Pen to facilitate downstream permeability processing and equilibrium labeling operations, and do not allow the sample to dry out during the course of the experiment;
e. Each sample is immersed in 0.2% Triton X-100 solution prepared in PBS and incubated for 5 min at room temperature for permeabilization;
f. Wash samples by submerging them 2-3 times in an open beaker containing PBS solutio
g. Gently remove excess liquid and carefully blot the liquid around the sample on the slide with filter paper. Keep the sample moist by placing the treated sample in a wet box.
2. DNase I treatment positive control experiment (optional step)
After sample permeabilization treatment, samples are treated with DNase I (recommended G3342) to prepare positive controls.
2.1. 100 μL of 1×DNase I Buffer (preparation: take 10 μL of 10×DNase I Buffer, then add 90 μL of deionized water and mix well) is added dropwise to the permeabilized samples and incubated for 5 min at room temperature;
2.2. Gently remove the excess liquid, add 100 μL of working solution containing DNase I (20 U/mL) (preparation method: take 10 μL of 10× DNase I Buffer, then add 2 μL of DNase I, and then add 88 μL of deionized water to mix), and incubate for 10 min at room temperature;
2.3. Gently remove excess liquid and wash the slides thoroughly 3-4 times in a staining vat filled with PBS.
Note: Positive control slides must be stained using a separate staining vat, otherwise residual DNase I on the positive control slides may introduce a high background on the experimental slides.
3. Marking and detection
3.1. Equilibration: Add 50 μL Equilibration Buffer to each sample to cover all the sample area and incubate for 10 min at room temperature;
3.2. Labeling solution preparation: Thaw EdUTP Labeling Mix and Equilibration Buffer on ice and mix enough TdT incubation buffer for all experiments according to the ratio of Recombinant TdT enzyme: EdUTP Labeling Mix: Equilibration Buffer=1 µL:5 µL:50 µL (1:5:50), the volume of reagents used in the specific experiments can be adjusted in equal proportions according to the size of the slides;
3.3. Negative control system: Prepare a control TdT incubation buffer without Recombinant TdT enzyme and replace it with ddH2O;
3.4. Labeling: Remove as much Equilibration Buffer as possible, then add 56 μL of TdT Incubation Buffer to each tissue sample and incubate at 37°C for 1 h; be careful not to dry the slides;
3.5. Wash the tissue samples immediately with PBS for 4 washes of 5 min each;
3.6. Click reaction: Remove the PBS buffer from the previous step, add 100 μL of click reaction solution dropwise on the sample to cover the sample, incubate for 30 min at room temperature away from light; (refer to the following table for the click reaction system, add each reagent in turn, mixing well while adding, and the amount of preparation can be increased or decreased proportionally, and it is recommended to prepare it beforehand)
|
Component |
Volume |
|
Reaction Buffer A |
925 μL |
|
Reaction Buffer B |
10 μL |
|
iF488 azide dye |
15 μL |
|
Reaction additive(Reagent C) |
50 μL |
|
Total volume |
1000 μL |
3.7. Remove the click reaction solution and immediately wash with PBS buffer 2-3 times for 5 min each;
3.8. Gently wipe off the PBS solution around the sample with filter paper;
3.9. Nuclear staining: Samples are stained in a staining vat, where slides are immersed in the dark into a staining vat containing DAPI solution (freshly prepared and diluted with PBS) and left at room temperature for 8 min (or nuclear staining is performed with Hoechst 33258);
3.10. Sealing: After the samples are stained, the tissue samples are washed three times with PBS for 5 min each time, then the excess liquid is gently removed and the samples are sealed with drops of anti-fluorescence quenching sealer (recommended G1401);
3.11. Microscopy: Immediately analyze the samples under a fluorescence microscope, slides are protected from light, DAPI stains both apoptotic and non-apoptotic cells blue, and only in the nuclei of apoptotic cells there is a green fluorescence localized by iF488 azide dye admixture.